What is ANT?
ANT (Atomistic NanoTransport) is a software package to compute the electrical current in atomically defined nanostructures. ANT combines self-consistent field electronic calculations (typically Density Functional Theory), Landauer transport and the (non-equilibrium) Greens functions formalisms.
At present, ANT software package is composed of the independent codes
- ANT.G
- ANT.1D
- ANT.U.

In ANT the target system is divided in three regions, see Figure 1:
- Left electrodes
- Device (scattering region)
- Right electrodes

Figure 1. Schematic representation of system set up; Left electrodes (L), device (D) and right electrodes (R).
ANT can compute the properties and phenomena needed for understanding the charge transport between bulk electrodes when these are connected by an atomic- or a molecular-size region and a bias voltage is applied between them, see Figure 2.
ANT provides:

Figure 1. Example of transport calculation performed by ANT.G. The conductance of a six unit-cell (8,0) CNT contacted to Au is computed. Palacios J. J. et. al. Phys. Rev. B. 77, 113403 (2008)
ANT.G, ANT.1D & ANT.U; DIFFERENCES AT A GLANCE
| MAIN FEATURES | WHAT’S FOR | DRAWBACKS | |
| ANT.G |
|
|
|
| ANT.1D |
|
|
|
| ANT.U |
|
|
Technical specifications
ANT.G
Code designed as a generic computational tool with application in nanoelectronics. It provides an excellent compromise between computational cost and electronic structure definition as long as the aim is to compare with experiments where the precise atomic structure of the electrodes is not relevant or defined with precision.
Straightforward use of ANT.G includes the computation of the zero-bias conductance (or, alternatively, the electrical current under an applied bias voltage) of a variety of nanoscale systems such as molecular bridges or simply metallic atomic contacts as those created with scanning tunneling microscope or break junction techniques. The use of ANT.G may be naturally extended to the computation of scanning tunnelling spectroscopy and electrostatic force microscopy.
Makes use of the embedded cluster approach, which is associated with the use of the parametrized Tight-Binding (TB) Bethe Lattice (BL).
Embedded cluster approach
In such approach the scattering region is described with the Density Functional Theory (DFT) while the electrodes are described with a lower level of accuracy through the above mentioned BT lattice. This approach is valid because, while the detailed atomic and electronic structure of the device region is crucial, farther away from the scattering region these become less important. However, consequently, it is necessary to include a sufficiently wide section of the bulk electrodes in the device region so that the interface region between electrodes and scattering region is described properly. The model is generated by connecting a site with N nearest-neighbors in directions that can be those of a particular crystalline lattice.
The fact that this method cannot describe precisely the atomic structure of the electrodes can become useful when comparing the results with real experiments where the exact atomic geometry of the electrodes is not known and cannot be controlled in the experiment.
ANT.G is designed to have a low computational cost, thus, it becomes an excellent tool when dealing with big systems or if you wan to have “fast” prelimirary results.
The technical specifications are:
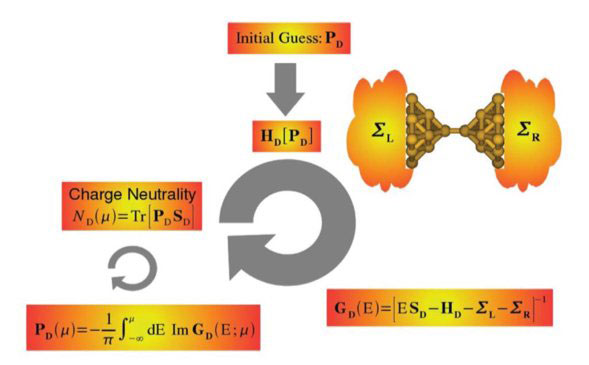
Figure 3. Diagram illustrating the self-consistent procedure for calculating the electronic structure in the embedded cluster approach as implemented in ANT.G. The central aspects of the used approach and of the one-body Green’s function and Landauer formalisms are given in D. Jacob and J. J. Palacios, J. Chem. Phys. 134,044118 (2011).
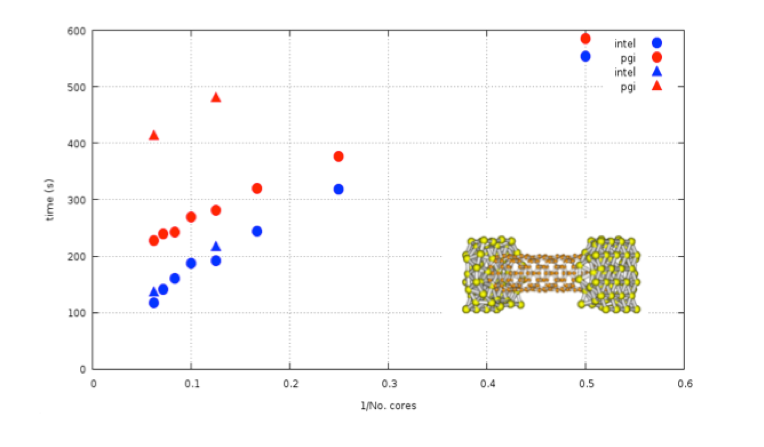
Figure 4. Performance of ANT.G in PGI and Intel compilers at low and high precision calculations
ANT.1D
Software specifically designed for the computation of the conductance at zero bias voltage in quasi-one-dimensional systems such as atomic chains, nanowires, carbon nanotubes, graphene nanoribbons, etc., which may present a disordered central region where scattering takes place. Other systems without an underlying one-dimensional symmetry such as molecular bridges can also be computed with ANT.1D by modeling the electrodes as finite-section quasi-one-dimensional wires.
ANT.1D is more demanding than ANT.G from the computational point of view, but presents the advantage of expanding the range of applicability of transport calculations to situations where the electrodes have a well-defined atomic structure.
ANT.1D has been tested with the gnu C++ compiler on various 32- and 64-bit platforms
Supercell approach
In this approach the electronic structure of the device region and the electrodes is calculated with ab-initio electronic structure codes for periodic systems that use localized basis sets such as CRSYTAL or SIESTA.
The system configuration of Left (L) anf Right (R) electrodes plus the central scattering or Device (D) region, see figure 5. In particular, the two leads L and R have to be connected where the electronic structure has relaxed to that of a bulk (i.e., infinite) nanowire thus, far enough away from the scattering region.
The technical specifications are:

Figure 4. Illustration of the super-cell approach to calculate the electronic structure of the device and of the leads: (a) One-dimensional periodic system to calculate the electronic structure of the device region. (b) and (c): Infinite nanowires to calculate the electronic structure of the left (L) and right (R) semi-infinite leads. (d) Sketch of the setup of the physical system: The device region (D) is suspended between two semi-infinite leads L and R. Further details of the method are given in D. Jacob and J. J. Palacios, J. Chem. Phys. 134,044118 (2011).
Basic functionalities of ANT.1D:
Advanced functionality for treating strong electronic correlations:
ANT.U
This code is specifically designed for the computation of the conductance at zero bias voltage in, e.g., graphene-based systems using a one-orbital minimal model and on-site interactions (Hubbard model).
ANT.U is an easy-to-use program for the study of spin transport in 1D systems (fundamentally graphene-based systems like nanoribbons and nanotubes) which applies the Landauer formalism to tight-binding Hamiltonians with a local Coulomb interaction (U).
The local Coulomb potential is obtained self-consistently using the mean-field Hubbard model.
Discover our Case Studies
Simulation is used for a wide variety of cases to solve materials-related challenges. Learn more about application areas.
Got any question ?
We are happy to hear from you. We adjust our support to meet the users requirements and proviide solutions to their specific problem.