WHAT IS ANT?
ANT (Atomistic Nano Transport) is a software package to compute the electrical current in atomically defined nanostructures.
ANT combines self-consistent field electronic calculations (typically Density Functional Theory), Landauer transport and the non-equilibrium Greens functions formalisms. At present, ANT software package is composed of the independent codes:
- ANT.G
- ANT.1D
- ANT.U
ANT.G is fully integrated into quantum chemistry code GAUSSIAN, while ANT.1D can be used as a post-procedure to a CRYSTAL calculation.
You can find a description of ANT and its main features HERE.
ANT.G
ANT.G interfaces with the quantum chemistry code GAUSSIAN03/09 to compute the electronic structure with open boundary conditions in equilibrium and non-equilibrium situations i.e., with zero and finite bias voltage. ANT follows an onion-like shell structure, modelling the far electrodes with a parametrized Bethe lattice, while computing the central scattering region at the DFT level.
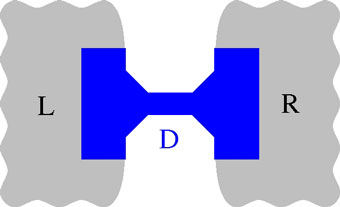
Schematic representation of system set up; Left electrodes (L), device (D) and right electrodes (R).
Basic functionality:
Spin-resolved transmission spectrum [T(E)] in equilibrium (linear response) or out of equilibrium (finite bias voltage) at zero temperature (T..dat).
Total density of states [DOS(E)] and DOS projected on selected atoms and orbitals (DOS..dat).
Orbital eigenchannel analysis (T.dat, t.dat, and standard output .log).
Average electronic potential on atoms (V..dat and standard output .log).
Mulliken and spin population analysis (Q.dat and standard output .log). When the DOS is requested, the DOS projected on atoms at a given energy is also dumped into Q.dat.
Advanced features:
Possibility of computation of electrostatic forces under an applied bias voltage.
Spin manipulation directives for the creation of self-consistent solutions with tailored spin textures.
Correlation effects at the Dynamical Mean Field Theory level.
Complementary codes
CRY2TB: Code for the extraction of Bethe lattice parameters from CRYSTAL calculations.
ANT MANUALS
Installing ANT.G
GAUSSIAN 03/09 input file
ANT.G input file
Bethe Lattice input file
TECHNICAL SPECIFICATIONS
ANT.G is written in FORTRAN90 and uses OpenMP directives to take full advantage of multiprocessor nodes.
ANT.G has been thoroughly tested with PGI compilers on various 32- and 64- bit platforms.
EXAMPLES
Gold: BL079.dat
Nickel: BL028.dat
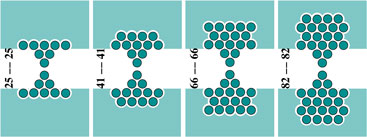
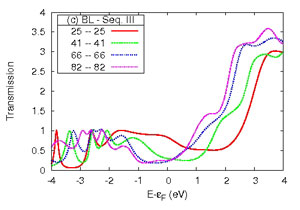
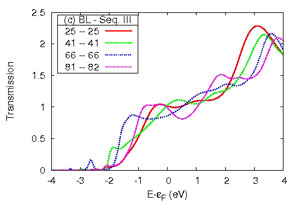
Transmission function or conductance of a series of Al (lower left) and Cu (lower right) nano-contacts of increasing atomic detail of the electrodes as shown in the upper panel.
CURRENT DEVELOPERS
J.J. Palacios, Departamento de Física de la Materia Condensada, Universidad Autónoma de Madrid, Spain
D. Jacob, Theory Department, Max-Planck-Institute for Microstructure Physics, Halle, Germany
María Soriano, Departamento de Física de la Materia Condensada, Universidad Autónoma de Madrid, Spain
FORMER DEVELOPERS
A. J. Perez-Jimenez, Departamento de Química-Física, Universidad de Alicante, Spain
E. Louis, Departamento de Física Aplicada, Universidad de Alicante, Spain
E. San Fabian, Departamento de Química-Física, Universidad de Alicante, Spain
J. A. Verges, Departamento de Física Teórica de la Materia Condensada, Instituto de Ciencia de Materiales de Madrid, CSIC, Spain
HOW TO CITE ANT:
If you are using ANT.G, please cite the appropriate references:
Fullerene-based molecular nanobridges: A first-principles study, J.J. Palacios, E. Louis, A.J. Perez-Jimenez, and J.A. Verges, Phys. Rev. B 64, 115411 (2001)
First-principles approach to electrical transport in atomic-scale nanostructures, J.J. Palacios, A.J. Perez-Jimenez, E. Louis, E. SanFabian, and J.A. Verges, Phys. Rev. B 66, 035322 (2002)
Molecular Electronics with GAUSSIAN98/03, J.J. Palacios, A.J. Perez-Jimenez, E. Louis, E. SanFabian, J.A. Verges, and Y. Garcia, Computational Chemistry: Review of Current Trends, Vol 9, 1 (2005)
Critical comparison of electrode models in density functional based quantum transport calculations, D. Jacob and J.J. Palacios, J. Chem. Phys. 134, 044118 (2011)
Specific References
Finite bias voltage: Implementing the Keldysh formalism into ab-initio methods for the calculation of quantum transport: Application to metallic nanocontacts, E. Louis, J.A. Verges, J.J. Palacios, A.J. Perez-Jimenez, and E. SanFabian, Phys. Rev. B 67, 155321 (2003)
Spin resolved transport and magnetism: Magnetic and orbital blocking in Ni nanocontacts, J.J. Palacios, D. Jacob, and J. Fernandez-Rossier, Phys. Rev. B 71, 2204303 (2005)
Back gate voltage and Fermi level control: Coulomb blockade in electron transport through a C60 molecule from first-principles, J.J. Palacios, Phys. Rev. B 72, 125424 (2005)
Orbital eigenchannel analysis: Orbital eigenchannel analysis for ab initio quantum transport calculations, D. Jacob and J.J. Palacios, Phys. Rev. B 73, 075429 (2006)
Correlated electronic transport: Kondo effect and conductance of nanocontacts with magnetic impurities, D. Jacob, K. Haule, and G. Kotliar, Phys. Rev. Lett. 103, 016803 (2009)
Dynamical Mean-Field Theory for Nano-Transport: Dynamical mean-field theory for molecular electronics…, D. Jacob, K. Haule, and G. Kotliar, Phys. Rev. B 82, 195115 (2010)
Other Publications
Metal contacts in carbon nanotube field-effect transistors: Beyond the Schottky barrier paradigm, J.J. Palacios, P. Tarakeshwar, and Dae M. Kim, Phys. Rev. B 77, 113403 (2008)
Electron transport properties of the porphyrin molecule located between gold electrodes, I. Yanov, Y. Kholod, J. Leszczynski, and J.J. Palacios, Chem. Phys. Lett. 445, 238 (2007)
Formation of a Metallic Contact: Jump to Contact Revisited, C. Untiedt, M.J. Caturla, M.R. Calvo, J.J. Palacios, R.C. Segers, and J.M. van Ruitenbeek, Phys. Rev. Lett. 98, 206801 (2007)
Transport in magnetically ordered Pt nanocontacts, J. Fernandez-Rossier, D. Jacob, C. Untiedt, and J.J. Palacios, Phys. Rev. B 72, 224418 (2005)
Ballistic resistivity in alluminium nanocontacts, A. Hasmy, A.J. Perez-Jimenez, J.J. Palacios, P. Garcia-Mochales, J.L. Costa-Kramer, M. Diaz, E. Medina, P.A. Serena, Phys. Rev. B 72, 245405 (2005)
Electronic transport and vibrational modes in a small molecular bridge: H2 in Pt nanocontacts, Y. Garcia, J.J. Palacios, A.J. Perez-Jimenez, E. Louis, E. SanFabian, and J.A. Verges, Phys. Rev. B 69, 041402 (2004)
Molecular electronics and first-principles methods, J.J. Palacios, A.J. Perez-Jimenez, E. Louis, E. SanFabian, and J.A. Verges, https://xxx.lanl.gov/abs/cond-mat/0301270
First-principles phase-coherent transport in nanotubes with realistic contacts, J.J. Palacios, A.J. Perez-Jimenez, E. Louis, E. SanFabian, and J.A. Verges, Phys. Rev. Lett. 90, 106801 (2003)
Analysis of scanning tunneling spectroscopy experiments from first principles: The test case of C60 adsorbed on Au(111), A.J. Perez-Jimenez, J.J. Palacios, E. Louis, E. SanFabian, and J.A. Verges, ChemPhysChem 4, 388 (2003)
An ab-initio approach to electrical transport in molecular devices, J.J. Palacios, E. Louis, A.J.. Perez-Jiménez, E. SanFabian, and J.A. Verges, Nanotechnology 13, 378 (2002)
Electronic transport through C60 molecules, J.J. Palacios, A.J. Perez-Jimenez, E. Louis, and J.A. Verges, Nanotechnology 12, 160 (2001)
ANT.1D
ANT.1D makes use of a pre-computed Hamiltonian for the scattering section and for the disorder-free regions. It can thus be used with tight-binding parametrized models or as a post-processing procedure to a one-dimensional periodic boundary conditions self-consistent electronic structure calculation using, e.g., CRYSTAL or SIESTA codes.
Basic functionality:
Spin-resolved coherent transmission spectrum and eigenchannel transmissions. Density of states and projected density of states on selected atomic orbitals. Charge and spin Mulliken population analysis.
Additional Features
Interface for extracting the necessary Hamiltonians from CRYSTAL calculations.
Advanced features:
Correlated electronic transmission function taking into account electronic interaction self-energy. Interface to NCA and OCA impurity solvers for calculating electronic self-energy. Dynamical Mean-Field Theory for nanoscopic conductors.
MANUAL
Download ANT.1D Manual
TECHNICAL ESPECIFICATIONS
ANT.1D is completely written in FORTRAN90 and has been parallelized with MPI. It has been tested with the Intel compiler on various 32- and 64-bit platforms with and without MPI. It also been tested with the Portland group compiler, but only without MPI. The interface with the CRYSTAL03 and CRYSTAL06 code is written in ANSI C++, and has been tested with the gnu C++ compiler on various 32- and 64-bit platforms.
EXAMPLES
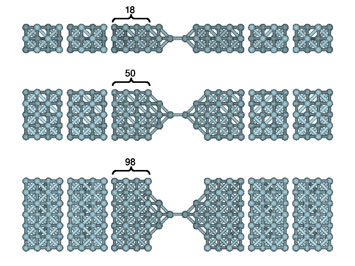
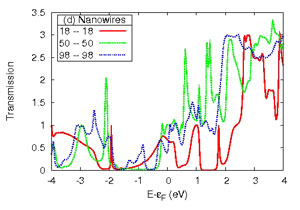
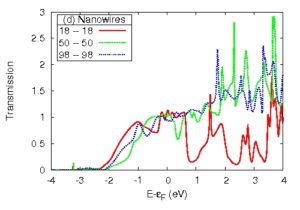
Transmission function or conductance of a series of Al (lower left) and Cu (lower right) nanocontacts for increasing section size of the nanowire electrodes as shown in the upper panel.
CURRENT DEVELOPER
D. Jacob, Theory Department, Max-Planck-Institute for Microstructure Physics, Halle, Germany
REFERENCES
If you are using ANT.1D, please cite the appropiate references:
Generic References
Critical comparison of electrode models in density functional based quantum transport calculations, D. Jacob and J.J. Palacios, J. Chem, Phys. 134, 044118 (2011)
Specific References
Correlated electronic transport: Kondo effect and conductance of nanocontacts with magnetic impurities, D. Jacob, K. Haule, and G. Kotliar, Phys. Rev. Lett. 103, 016803 (2009)
Spin-orbit coupling in coherent transport: Anisotropic magnetoresistance in nanocontacts, D. Jacob, J. Fernandez-Rossier, J.J. Palacios, Phys. Rev. B 77, 165412 (2008)
Dynamical Mean-Field Theory for Nano-Transport: Dynamical mean-field theory for molecular electronics…, D. Jacob, K. Haule, and G. Kotliar, Phys, Rev. B 82, 195115 (2010)
Coherent transport calculations of graphene nanoribbons: Coherent transport in graphene nanoconstrictions, F. Munoz-Rojas, D. Jacob, J. Fernandez-Rossier, J.J. Palacios, Phys. Rev. B 74, 195417 (2006)
Spin Revolved Transport and Magenism: Magnetic and orbital blocking in Ni nanocontacts, D. Jacob, J.J. Palacios, and J. Fernandez-Rossier, Phys. Rev. B 71, 220403 (2005)
Orbital eigenchannel analysis: Orbital eigenchannel analysis for ab initio quantum transport calculations, D. Jacob and J.J. Palacios, Phys, Rev. B 73, 075429 (2006)
ANT.U
This code is specifically designed for the computation of the conductance at zero bias voltage in, e.g., graphene-based systems using a one-orbital minimal model and on-site interactions (Hubbard model).
ANT.U is an easy-to-use program for the study of spin transport in 1D systems (fundamentally graphene-based systems like nanoribbons and nanotubes) which applies the Landauer formalism to tight-binding Hamiltonians with a local Coulomb interaction (U).
The local Coulomb potential is obtained self-consistently using the mean-field Hubbard model.
Do you have a question ?
Contact us. SIMUNE Team is happy to hear from you.